Ce este sindromul Prader-Willi?
Sindromul Prader-Willi (PWS) este o tulburare genetică care apare la aproximativ una din fiecare 15.000 de nașteri. PWS afectează bărbați și femei cu o frecvență egală și afectează toate rasele și etniile. PWS este recunoscută ca fiind cea mai frecventă cauză genetică a obezității care amenință viața copiilor.
Sindromul a fost descris pentru prima dată de către doctorii elvețieni Andrea Prader, Alexis Labhart și Heinrich Willi în 1956, pe baza caracteristicilor clinice ale celor nouă copii pe care i-au examinat. Caracteristicile comune definite în raportul inițial includea mâinile și picioarele mici, creșterea anormală și compoziția corporală (statură mică, masa corporală foarte scăzută și obezitatea cu debut precoce), hipotonia (mușchii slabi) la naștere, foamea permanentă, obezitatea extremă și retard intelectual.
PWS provine dintr-o anomalie a cromozomului 15, iar diagnosticul definitiv se bazează acum pe testarea genetică.
Care sunt simptomele sindromului Prader-Willi?
Simptomele sindromului Prader-Willi sunt probabil datorate disfuncției unei porțiuni a creierului numită hipotalamus. Hipotalamusul este un mic organ endocrin la baza creierului care joacă un rol crucial în numeroase funcții ale corpului, incluzând reglarea foamei și a sațietății, a temperaturii corpului, a durerii, a reglării somnului și a fluidelor, a emoțiilor și a fertilității. Deși se crede că disfuncția hipotalamică conduce la simptomele PWS, nu este clar modul în care anomaliile genetice cauzează disfuncția hipotalamică.
Simptomele PWS se schimbă de-a lungul timpului la persoanele cu PWS. În general, există două etape ale simptomelor asociate cu PWS:
Începutul vieții
Sugarii cu PWS sunt hipotonici sau „floppy”, cu tonus muscular foarte scăzut. Un plânset slab și un reflex de supt slab sunt tipice. Bebelușii cu PWS, de obicei, nu pot fi alăptați și necesită frecvent hrănirea cu tub nazogastric. Acești sugari pot suferi de „eșecul de a progresa” dacă dificultățile de hrănire nu sunt atent monitorizate și tratate. Pe măsură ce acești copii cresc, puterea și tonul muscular se îmbunătățesc în general. Dezvoltările motorii sunt atinse, dar de obicei sunt întârziate. Copiii mici intră, de regulă, într-o perioadă în care pot începe să câștige greutate cu ușurință, înainte de a avea un interes sporit față de alimente.
Copilărie și perioadele următoare
Un apetit neregulat și o creștere ușoară în greutate caracterizează etapele ulterioare ale PWS. Aceste caracteristici încep cel mai frecvent între vârsta de 3 și 8 ani, dar sunt variabile în debut și intensitate. Persoanele cu PWS nu cunosc indicii normale de foame și de sațietate. De obicei, aceștia nu sunt capabili să controleze consumul de alimente și vor mânca prea mult dacă nu sunt supravegheați îndeaproape. Interesul crescut pentru hrană este foarte frecvent . În plus, rata metabolică a persoanelor cu PWS este mai mică decât în mod normal. Lăsate netratate, această combinație de probleme conduce la obezitate morbidă și la numeroase complicații.
În plus față de obezitate, o varietate de alte simptome pot fi asociate cu sindromul Prader-Willi. Indivizii prezintă, de obicei, provocări cognitive, cu valori ale măsurătorilor de inteligență (IQ), de la insuficiență intelectuală mică, normală până la moderată. Cei cu IQ normal prezintă, de obicei, dificultăți de învățare. Alte probleme pot include deficiența hormonului de creștere / statura scurtă, mâinile și picioarele mici, scolioza, tulburările de somn cu somnolență excesivă în timpul zilei, pragul de durere ridicat, apraxia / dispraxia vocală și infertilitatea. Dificultățile comportamentale pot include simptome obsesiv-compulsive, ciupitul pielii și dificultăți în controlul emotiilor. Adulții cu PWS prezintă un risc crescut de boală mintală. PWS este un spectru și simptomele variază în funcție de gravitate în rândul indivizilor.
Ce cauzează sindromul Prader-Willi?
PWS este cauzat de lipsa materialului genetic activ într-o anumită regiune a cromozomului 15 (15q11-q13). În mod normal, persoanele moștenesc o copie a cromozomului 15 de la mama lor și de la tatăl lor. Genele din regiunea PWS sunt în mod normal active doar pe cromozomul venit de la tată. În PWS, defectul genetic care cauzează inactivitatea cromozomului 15 de la tată (cromozomul paternal 15) poate să apară în unul din trei moduri:
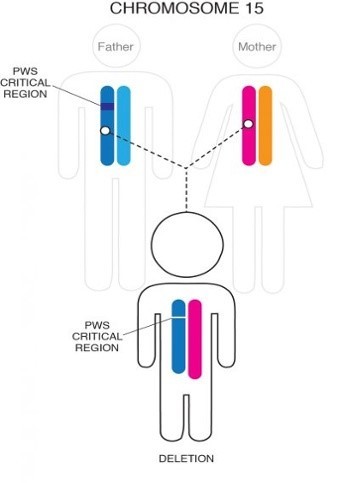
PWS prin deleție
Cel mai adesea, o parte din cromozomul 15 care a fost moștenit de la tatăl persoanei lipsește sau este șters în această regiune critică. Această deleție mică apare în aproximativ 70% din cazuri și, de obicei, nu este detectabilă cu analize genetice de rutină, cum ar fi amniocenteza.
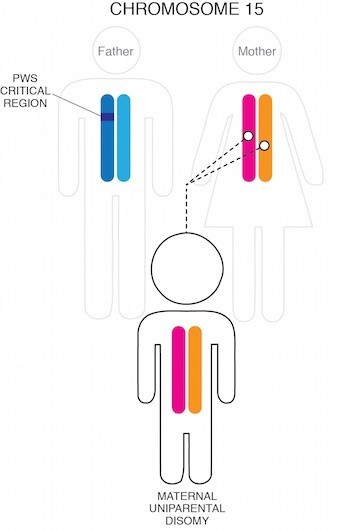
PWS prin UPD
Alte aproximativ 30% dintre cazuri apar atunci când o persoană moștenește doi cromozomi 15 de la mamă și niciunul de la tatăl lor. Acest scenariu se numește disomie uniparentală (UPD).
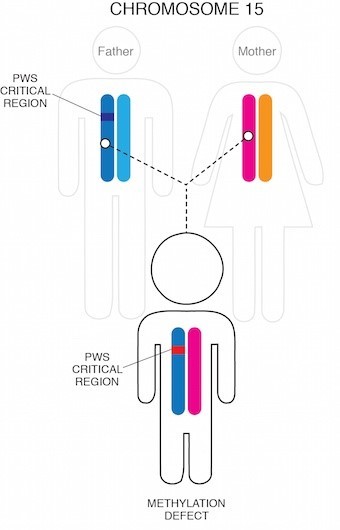
PWS prin imprinting
În cele din urmă, într-un procent foarte mic de cazuri (1-3%), o mică mutație genetică în regiunea Prader-Willi este determinat de inactivitatea cromozomului paternal 15 (chiar dacă este prezent).
Cum generează aceste defecte genetice simptomele observate în sindromul Prader-Willi?
Regiunea PWS a cromozomului 15 este una dintre cele mai complexe regiuni ale genomului uman. Deși s-au înregistrat progrese semnificative în înțelegerea și caracterizarea modificărilor genetice asociate cu PWS, mecanismul exact prin care lipsa materialului genetic funcțional în această regiune duce la simptomele asociate cu PWS nu este înțeleasă. Oamenii de știință studiază în mod activ rolul normal al secvențelor genetice din regiunea PWS și modul în care pierderea lor afectează hipotalamusul și alte sisteme din organism.
Există diferențe în severitatea PWS pe baza subtipului genetic?
Pot exista anumite diferențe subtile în ceea ce privește caracteristicile PWS pe baza subtipului genetic: de exemplu, cei cu ștergere pot fi jupuți cu părul ușor în comparație cu ceilalți membri ai familiei și pot fi mai susceptibili la crize convulsive; cei cu PWS de UPD pot prezenta un risc mai mare de boală mintală la vârsta adultă. În ansamblu, totuși, există o suprapunere considerabilă între diferitele subtipuri genetice. Este posibil ca mii de gene din afara regiunii PWS, care prezintă variații normale între indivizi, să contribuie, de asemenea, semnificativ la variabilitatea simptomelor PWS între cei cu tulburare.
Cum este diagnosticat PWS?
PWS este diagnosticat printr-un test de sânge care caută anomalii genetice specifice PWS – numită „analiză de metilare”; testul FISH (hibridizare fluorescentă in situ) identifică PWS prin deleție, dar nu diagnostichează alte forme de PWS. Testul de metilare va identifica toate tipurile de PWS și este testul preferat pentru diagnosticare. Dacă se efectuează mai întâi un test de metilare, pot fi necesare teste suplimentare pentru a determina dacă PWS este cauzată de o deleție paternă, UPD sau o mutație de imprimare. În cazurile în care se suspectează o mutație de imprimare, sângele poate fi, de asemenea, extras de la părinți.
Este ereditar sindromul Prader-Willi?
Deleția și UPD sunt întâmplătoare și, în general, nu sunt asociate cu un risc crescut de reapariție la sarcini ulterioare. În cazul imprintingului, sindromul Prader-Willi poate să reapară în cadrul unei familii. Familiile care se confruntă cu riscul pentru PWS ar trebui să vorbească cu un consilier genetic.
Există un tratament pentru sindromul Prader-Willi?
În prezent, nu există nici un tratament pentru sindromul Prader-Willi, iar majoritatea cercetărilor până în prezent au vizat tratarea simptomelor specifice. Pentru mulți indivizi afectați de tulburare, eliminarea unora dintre cele mai dificile aspecte ale sindromului, cum ar fi apetitul insațiabil și obezitatea, ar reprezenta o îmbunătățire semnificativă a calității vieții și a capacității de a trăi independent.
Notă: această prezentare reprezintă traducerea unui extras preluat de pe site-ul Foundation for Prader-Willi Research ce poate fi găsit aici